Polymerase Chain Reaction (PCR) is a powerful technique for detecting the presence of nucleic
acids of specific sequence - that is, DNA and RNA: polymers that can be viewed as long 'sentences' in an alphabet of four different 'letters'. By running through cycles of sequence
duplication, the quantity of the target sequence approximately doubles with each step,
generating sufficient from small initial quantities to be analysed by chemical
techniques too insensitive to detect the original quantity. Additionally,
because it uses sequence-specific ‘primers’ to start the ball rolling, careful
design of these permits a very high degree of sequence specificity.
During the Coronavirus pandemic, much internet chatter and
mythology has arisen around this technique, which this article will attempt to address.
PCR - a rough guide.
PCR was invented by Kary Mullis, a chemist at Cetus
Corporation, in 1983. This invention earned him the Nobel Prize in Chemistry in
1993.
In biology, the deoxygenated form of nucleic acid, DNA, typically exists as double
strands – the famed Double Helix. The shape of
one strand is ‘complementary’ to that of the other; they fit neatly together
and stabilise each other. This complementarity also allows the strands to
specify each other; to form each other's template.
Prise apart a double strand and each single strand has all the information required to recreate its complement, enabling us to create two copies of each helix from one, iteratively. This potential for doubling leads to exponential increase, when unconstrained.
A useful visualisation of a DNA double strand is to imagine two
rows of people facing each other, shaking hands. If we add a rule that a man on
one row has to be opposite a woman on the other – no same-sex pairings – we
have a reasonable approximation of the structure of DNA. If we separated the two
rows, unclasping their hands, we have all the information we need to create two identically-constituted copies of the original double row.
Wherever there is a man, we know we can only add a woman, and vice versa.
These facing lines
have opposite directionality – each left shoulder faces the right shoulder on
the other line, and vice versa. And so it is with DNA, with the added
constraint that the process of reconstitution of the complement can only happen sequentially, and in one direction:
‘left-to-right’, never ‘right-to-left’, so starting from the other 'end' on each strand.
Here’s what happens in PCR:
- The DNA strands are separated using heat.
- Short primer sequences of DNA bind to specific
targets on the separated strands. Two different primers are used, one for each strand,
bracketing whatever lies between.
- An enzyme, DNA polymerase, rebuilds the complementary strand, starting at the bound
primer on each strand and working ‘left-to-right’ from each primer site, such that each acts in the opposite direction with respect to the other, and they cover the same segment of helix.
- The extension of the new strand from each primer
binding site eventually passes through the primer binding sequence of the opposite segment,
and so we have neatly duplicated everything bracketed by the primer binding
sequences plus the primer binding sequences themselves.
- Go to 1.
This is a ‘cycle’. In Mullis’s original design, fresh DNA
polymerase had to be added each cycle, rendering it quite laborious and more prone to contamination.
Additionally, it was necessary to pick a number of cycles and then analyse the outcome, to gain a coarse yes/no answer on the presence or absence of the pair of
primer binding sequences. Two enhancements have enabled automation and
quantification. First, a heat-stable DNA polymerase from a microorganism
inhabiting hot springs is used (co-discovered by Hudson Freeze!), avoiding the need to add more each cycle.
Secondly, sequence-specific chemical markers (probes) are added
that allow determination of the cycle at which a positive signal is found. These must lie between the primers; as amplification proceeds, more of the probe is bound. Being an exponential process, there is a lag period where doubling does not
make a great difference to overall quantity – 2, 4, 8, 16, 32 etc. But as this
progresses, there is quite a sharp boundary where the quantity rises very
rapidly above the threshold of detection - similar to, for similar reasons, the rapid onset of illness after a few days' unnoticed incubation. This is the Ct threshold, the cycle
at which clear signal emerges. It is not the maximum number of cycles for which
the cycler runs.
Because the coronavirus genome is RNA, an additional first step is
required, to convert the RNA sequence into a double-stranded DNA copy, which
can then be amplified in the normal way. This is the ‘RT’ of RT-PCR: conversion
from RNA to DNA is ‘Reverse Transcription’ (although confusingly, RT is sometimes used to signify 'Real Time').
The extreme sensitivity of PCR is due to this exponential
doubling; its very high specificity is due to the ability to design primers
that are unique to the genome of interest. It's like using paired
literature ‘primers’: “It is a truth universally acknowledged” and “want of a
wife”. Armed with these primers, one could go ‘fishing’ in a library and pull
out copies of Pride and Prejudice with high accuracy. But they may also pull out
books of notable quotations, or torn-out first pages. It is not a guarantee of completeness,
and may misidentify sequence that has been recombined into other settings. But
what it won’t do is find something that isn’t there. Both primer sequences must
be present, they must be on opposite strands and 'downstream' of each other, and the probe sequence must lie between them. This sequence combination creates a high degree of specificity.
The Myths.
Many false claims are made about PCR, particularly among
‘Covid deniers’ – those who think the disease does not even exist, and other
illnesses or causes of death have been routinely misattributed.
1) "Nobel Prize Winning Inventor of PCR Kary Mullis
says it should not be used for diagnosis”.
a. I can find no evidence of him actually saying
this. You're usually directed to a video of him discussing hypersensitivity in relation to HIV/AIDS, a link
he denied, but he does not actually make the attributed comment in that clip. This is a common
theme when people try and import experts on subjects about which they
themselves are vague. It’s not what the expert says, but what they think he says. Many people effectively argue that he has said that the test he invented is useless - that
it will detect things that are not there if you run enough cycles. He doesn’t
say this, and it is odd to bring the inventor in as an authority to blow his
own invention out of the water!
b.
Mullis’s Nobel was for chemistry. He had no
authority on clinical matters. Again, this keys in to lay understandings of
expertise: a Nobel laureate is assumed an expert way beyond his field. Sometimes, by the laureate themselves.
c.
His Nobel citation included its use in diagnosis as one of
the main reasons for getting the Prize. He did not say 'hang on a sec...". He also filed a patent for diagnostic use, and co-authored at least one paper on that theme.
d.
The inventor of a technology has no primacy of
opinion on its applications. Tim Berners-Lee doesn't tell us what the internet can be used for.
e.
Whatever he thought, PCR is routinely used for
diagnosis, and has been for 25 years or more. A quick Google reveals numerous examples: tick-borne diseases of cattle in Maharashtra
State, a virus of orchids, detection of bacteria in cerebrospinal fluids,
influenza surveillance ... so regardless, clinicians are happy to rely
on it, and that’s good enough for me!
Are we seriously to suppose generations of scientists and doctors blind to the lack of utility of PCR, on the basis of a 10 second mumble by its inventor?
2)
“NPWIOPCRKM says quantitative PCR is an
oxymoron”.
a.
Again,there is no evidence he actually said
this. The quote is in a 1996 article by
John Lauritsen,
on HIV/AIDS and the gay community, but the origin of the attributed quote is obscure, and may simply be a paraphrase of Lauritsen's understanding of Mullis's position.
b.
Whatever he thought 25 years ago,
quantitative PCR is a thing. It is used routinely, for example, in clinical
management of hepatitis and – ironically -
HIV.
Look at Figure 1 in the linked paper:
Each graph represents a set of serial dilutions of a known starting quantity of viral RNA, for the different genes that each primer pair targets, with a very good agreement of data points to the best-fit line. There is a clear linear relation between
the Ct threshold at which signal is detected and viral RNA concentration. This is
quantitative, not a simple single-point yes/no. But note also that there is not a consistent Ct corresponding to a given number of sequence copies. There are 5 Ct values (hence 5 'doublings') between the highest and lowest, in these graphs, for the same amount of nucleic acid. When people say "Ct should be x or lower", they demonstrate lack of awareness of this technological fact.
3) "They run for so many cycles they can find anything".
This is related to 2. People saying this assume that the process runs to completion before inspection. They typically quote a cycle count in excess of 40, which is getting towards the limit of usefulness but, as can be seen above, modern methods do not simply run to max then peek. They monitor progress in real time, and can tell precisely when the detection threshold is passed. Signal generally appears well below the 40th cycle - see the y axes in the graphs above. Mullis himself published a paper utilising 60 cycles. And, however long you run, you need at least one correctly-oriented pair of your primer binding sequences to get any amplification at all. It does not conjure up positives from thin air.
Note that in the graphs above the 40-cycle points sit neatly on the best-fit line. They aren't going haywire due to amplification of non-existent sequence, as one would expect to start seeing at high Ct if this were an issue. Pretending for a moment that Ct was the end-point cycle count, then if Ct40 routinely gave false positives, phantom amplification of non-existent sequence should add to the amplification of the real sequence, pushing Ct40 points off the line drawn through lower-Ct points. The log-linear relationship with starting concentration should break down.
Running PCR takes resource - people, time, machines and reagents. To optimise throughput, labs frequently pool N subsamples and run one PCR on the pool. If it comes out negative, they have saved running N-1 individual PCRs. If it's positive, they test the reserved portions of the pool individually. The optimum N depends on prevalence - there's no point pooling at such a high N that every pool contains a positive. Nor should N be so high that true positives are diluted to undetectable levels. Nonetheless, because the pooling inevitably dilutes each member by a factor of N, they may use a higher Ct threshold for a pool-positive than an individual-positive - it would push dilution factors to the left in the above graphs; N=10 would give 1 increment to the left on the x axis log scale, and hence report at the correspondingly higher Ct. Clearly, if incidental false-positives were an issue at high Ct, this attempt to optimise throughput would be entirely self-defeating; too many pools would need individual examination, because you'd pushed Ct into the supposed 'false-positive' region. But also note that this pooling will mask low-count positives, by dilution. The use of pooling is at odds with the belief that fraudulent Ct-hacking is in operation. Surely it would be easier to lie?
4)
“PCR cannot tell if you are infectious”
a.
Partially true; PCR does not categorically distinguish
live virus from viral debris containing the primer sequences. However, using one of
the quantitative methods, rather than the simple yes/no of Mullis’s day, a
degree of confidence in infectiousness can be provided. During an active
infection, viral load will be maximal early in the infectious period, as
replication adds RNA copies and the innate immune system fails to remove them at a
sufficient rate. In the post-viral phase, viral replication stops, and the amount
of false-positive non-viable debris will decrease by degradation, with no
replenishment. This predicts a relationship between active viral load and the
Ct threshold of detection, and this has been borne out by
comparing PCR results to a culture test of virus replication capacity.

As Ct value increases - meaning less viral RNA, whether whole or fragmentary - the chance of culture positivity declines quite rapidly after an initial lag. Viable virus is typically not detected beyond day 10 of infection, informing the current isolation rules. Ct cycles
of 35 or below would seem to be a reliable indicator of infectiousness, on this evidence.
b.
This lack of distinction may not matter anyway, depending on what action
the test informs. For clinical isolation and ward management, it is necessary to know if
there is active virus. But for case management, a clinician cares less that an
individual is shedding live virus; if they are symptomatically ill and PCR positive, even at Ct
values suggesting no active virus, they still know they have a case of ‘Covid’
to deal with. The illness does not stop when virus stops replicating. If only.
5)
“PCR cannot distinguish Covid from cold viruses
or flu”.
a.
This is untrue. PCR is one of the most powerfully specific
techniques available. The choice of primers needs to target sequences unique to
the ‘organism’ of choice; there is little sequence commonality between
coronaviruses and rhinoviruses, adenoviruses or influenza viruses, and all you need do is avoid that in choosing primers and probes. Due to
closer genetic relationship, there is some possibility of detecting other
coronaviruses – some primer targets cannot distinguish between SARS-CoV-1 and
SARS-CoV-2, if the binding sequences are common to both. But again, it is a simple
matter of using some other part of the viral genome. In the graphs above (2c), at least 5 different parts of the viral genome were covered by the primer sets, and some kits target 2 or 3 different genes simultaneously for added coverage. These kits were able to distinguish SARS-CoV-2 from other coronaviruses
SARS1, MERS, NL63, OC43, and 229E for all except the E
(Envelope) gene in some kits. Simply choosing a different target would
eliminate this, but given the comparative rarity of SARS1, it is unlikely that
many such false positives would occur in any case in a real-world setting. If they wished to pursue this cross-specificity angle, Covid deniers would then need to decide whether it is a pandemic of SARS2 or SARS1. Take your pick; it's definitely a pandemic of something!
b.
Covid sceptics who make this
claim were enthused by the US FDA’s recommendation that Covid kits that distinguish influenza
and Covid should be preferred in place of a simplex kit. “We
were right all along, it can’t tell them apart!”. But this misinterprets what
was said. The FDA were not saying that flu will cause false Covid
positives in a Covid-specific kit, but that those kits will not react to
influenza at all. Therefore, given
the advent of flu season, ‘multiplex’ tests,
that pick up either pathogen by having primers and markers for both, should be
used.
c. Take a look at
this 2018 (hence, pre-Covid) paper on detection of bacteria causing 'atypical pneumonias'. Common pneumonia-causing organisms are
viruses such as influenza and bacterial pathogens like Streptococcus pneumoniae and
Haemophilus influenzae. It is vital to distinguish bacterial and viral pneumonias since the latter will not respond to antibiotics. But the 'atypical' ones, although bacterial, will not respond to penicillin. It's clearly important to get this right, then, and absurd to argue that clinicians would settle for an inadequate test that routinely misguides treatment. The tests perfectly distinguished all 7 of the atypical pneumonia-causing bacteria, while not giving a false positive for any of a panel of common organisms - even at Ct40.
6)
“PCR generates [insert ridiculous percentage
here] false positives”.
False positives can certainly be a problem. They can overestimate prevalence when rare. At individual level, if a false-positive patient is incorrectly assigned to a room with Covid patients they are exposed to unnecessary infection risk. People may be unnecessarily worried by a potentially lethal disease, while in the community they may mistakenly assume they have infection-acquired immunity.
Test performance depends on sensitivity - the percentage of tests detecting virus when present - and specificity - the percentage of tests correctly reporting viral absence. A 99% sensitivity means there would be 1% false negatives. Likewise, 99% specificity gives 1% false positives. When randomly testing large numbers of people when the disease is at low level, most positives will be false. At the extreme, if nobody has Covid and you test 1,000 people, you get 10 false positives: 0% true. But clearly, the proportion of false positives to true is highly conditional upon the prior probability of having Covid, ie on prevalence, so unqualified statements like the above are misleading. If 1 in 50 has Covid and you test 1,000, you get 20 true positives to 10 false, ie 66% are true, not zero, for the same baseline specificity. Even this only applies for random testing. If people present for testing due to Covid-like symptoms, or contact with a positive, the prior probability of Covid goes up among those tested, and hence so does the proportion of true to false positives.
In ideal lab conditions, the analytic performance of these tests is typically near 100% on both measures, given a sufficient quantity of starting sequence (from c500 copies/ml). However, outside of the careful lab work undertaken when evaluating PCR, systematic errors can creep in. Contamination can occur at several stages, as can clerical error. The specificity drops from the optimal ~100% to about 99.5% in practice, giving a false positive rate of c0.5%. A 0.5% false positive rate is not nothing - 5 in 1000. But note that, if Covid is rare, this in itself will reduce the systematic error due to contamination - fewer people in the handling chain would be expected to have SARS-CoV-2 RNA about their persons.
If there's a lot of contamination, that itself indicates widespread virus.
7) "Most 'cases' are something else".
This integrates the arguments in 5) and 6). At the extreme, it would have every single 'Covid death' as a misattribution. This can't sensibly be the case, however. This argument implicitly holds that '28-day positive PCR' and 'deaths on a given day' are wholly independent, uncorrelated sets, since if every single 'case' were a false positive, each of them is statistically unlikely to die of any cause within the next 28 days. They have the same likelihood as the population as a whole, and any association is therefore random.
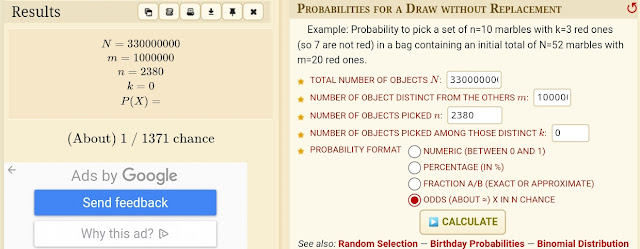
Let's examine this probabilistically. If the set of false positives and the set of deaths are completely independent, we can use simple
'draw x from y' probability calculations to determine the probability of intersect: the likelihood that a given proportion of people dying on a given day intersects with the set of 'false cases' in the previous 28.
Suppose there have been 30,000 cases a day over the last 28 days, or 840,000 total. Let's take a high-ish figure of 2000 for daily deaths, 100 of these attributed to Covid, and a population of 60 million. What is the probability that 100 out of 2000 draws from this population will have an uncorrelated characteristic shared by 840,000 random people? To express it in the terms of the calculator linked above, there are 60 million 'marbles' in total, 840,000 of them are red, and we want the probability you will find 100 red ones in 2000 draws without replacement (because you only die once!):

The calculator returns a probability of 1.04 x 10-26 for this; vanishingly small. For comparison, values in the region of the expected value of 28 out of 2000 'reds' - 28 incidental false positives - have a probability of 7.5%. 95% of the time we would expect a value between 19 and 37. Therefore, numbers of the order of 100/2000 are highly unlikely to occur with uncorrelated sets. 'Something' renders a PCR positive more likely to die within 28 days, or conversely, renders the deceased more likely to have had a recent positive test than random chance would predict. Whatever other cause you may prefer for this correlation, there clearly is one!
8)
“PCR determines what goes on the death
certificate. Fall off a ladder, you’re a Covid death”
This is not the case. Many countries do
conduct a simple intersect of two data sets, any-cause deaths and Covid tests within a
period (the last 28 days in the UK) in order to present daily headline figures.
But this rough-and-ready rule does not inform death certificates. A positive Covid test is
recorded if it is felt, by the clinician, to be a contributory factor, but not
otherwise. There are clear clinical signs of Covid, both pre and post mortem, and
these will inform the doctor recording the death. The perception seems to be
that doctors have suddenly lost the ability to diagnose, and rely solely on a
lab test. If there was a comorbidity - say obesity, hypertension, or diabetes - deniers believe that that, and not Covid, was the direct cause of death. Even if the illness was distinctively 'Covid-like', with classical clinical signs and biomarkers associated with infectious disease, and not at all like the deaths more usually associated with the comorbidity.
In fact, since not everyone who dies of Covid is tested, and some take longer than 28 days to die, the death-certificate figures are significantly higher than the 28-day figures, even after ruling out the miscounted car crashes etc.
There has been a bizarre rash of Freedom of Information requests in the UK, demanding to know how many deaths had Covid as 'the only cause'. Puzzled responders give counts of deaths with no known comorbidity, which of course are low, and don't actually answer the rather meaningless question. These low figures are then gleefully amplified as evidence that hardly anyone dies 'of' Covid. Instead, we have mysterious spikes of people dying 'of' their comorbidity!
9) "PCR doesn't work, but [therapeutic x] does".
Without
taking a position on the ivermectin/HCQ debate, we can see that the
co-occurrence of these two viewpoints is inconsistent. In order to both
evaluate a treatment, and to determine a suitable case for treatment,
there needs to be a means of distinguishing the disease from others
causing similar symptoms. If not PCR, what? Most papers touted as
supportive of ivermectin used PCR to identify cases. If PCR could not
distinguish SARS from flu or colds, it would not have been an
appropriate tool for this work. If you don't approve the method, you've
no entitlement to the results! While if ivermectin or HCQ are
Covid-specific, it's no good prescribing it for a cold or flu.
10)
“Kary Mullis (died August 2019) was killed for
his views on PCR”.
Get a grip.



The primer sequences are typically DNA, not RNA for PCR.
ReplyDeleteThanks for that; I will correct. I had in mind that DNA pol was RNA-dependent, but it appears to be so only in vivo.
Delete